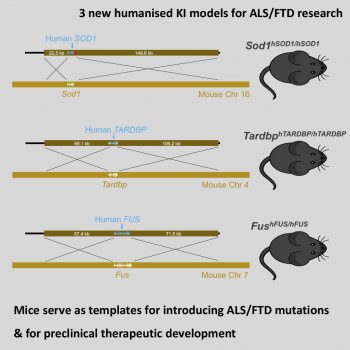
Generation of the first genomically humanised mice in which human genes for ALS/FTD-associated genes replace their mouse orthologues
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease characterised by progressive loss of muscle control, leading to spreading paralysis and death typically within five years of diagnosis. Understanding of the disease is limited and it is essentially untreatable. New mouse models of the disease could do much for the development of future therapies. A team of researchers at MRC Harwell, the Queen Square Motor Neuron Disease Centre at the UCL Queen Square Institute of Neurology, and the Canary Islands University Hospital, led by Thomas Cunningham, Elizabeth Fisher, and Abraham Acevedo-Arozena, have published a paper in iScience that describes their work to generate the first genomically humanised mice for 3 genes associated with ALS and frontotemporal dementia (FTD).
Although most ALS has an unknown cause, it can also be inherited and a number of genetic mutations have been identified as the source of this inheritance. These include the genes for the enzyme SOD1 and the RNA binding proteins FUS and TDP-43. Indeed, even in the absence of mutations, FUS and TDP-43 form abnormal pathological aggregates in the neurons of patients with ALS and FTD. A number of mechanisms for how mutations lead to disease have been proposed, but more work is needed.
Transgenic mice in which human ALS/FTD genes have been inserted have been of great use as they recapitulate human biochemistry and enable researchers to model these degenerative diseases within a short timeframe. However, these models are produced from random insertion of the human genes, meaning that effects seen in the mice could be due to the human protein or a negative effect of the disruption to the mouse gene sequence. Another issue is that this system usually results in insertion of multiple copies of the same gene, which is likely to increase the amount of that protein produced, which might also cause effects unrelated to the disease. Another option for creation of mouse models has been to edit mouse genes in order to insert equivalent mutations of interest, but this option is also limited where mouse and human proteins differ to too great an extent.
To address these limitations, the team generated three genomically humanised mouse models in which the human genes that code for SOD1, FUS, and TDP-43 precisely replaced their mouse counterparts. These human genes are driven by the mouse promoter to maintain normal expression levels and include the entire human sequence, including introns, to ensure that human splice variants can be produced.
The generation of these mice is technically challenging and required extensive quality control to assess that the human genes were inserted correctly and that human, not mouse, protein was produced within a physiological range. To demonstrate the capacity for this mouse to provide a useful model for the study of disease, the team then performed extensive characterisations of mice expressing the human FUS protein and demonstrated that these mice are normal throughout their lifespan. This means that human FUS can functionally replace the mouse version without detrimental effects.
The mice generated in this study carry healthy versions of human genes associated with ALS and FTD. It is then envisaged that mutations could be inserted using CRISPR/Cas9 so that it would be possible to generate a mouse model of disease and have a useful control mouse with the healthy human version of the protein. This would provide a more refined system for studying disease, thereby contributing to our commitment to the 3Rs. This system could be of great use to researchers wanting to better understand these diseases and develop new treatments. To support this, these lines are being made freely available to the scientific community through the European Mouse Mutant Archive, of which the MRC Harwell Archive acts as the UK node. This work also serves as a proof of principle, providing a pipeline through which more genomically humanised mouse lines could be produced in order to provide better models of human disease.
This work was supported by UKRI MRC, the Motor Neurone Disease Association, the ISCiii, CIBERNED, and the Rosetrees Trust.
Devoy, A, Price, G, De Giorgio, F, Bunton-Stasyshyn, R, Thompson, D, Gasco, S, Allan, A, Codner, GF, Nair, RR, Tibbit, C, McLeod, R, Ali, Z, Noda, J, Marrero-Gagliardi, A, Brito-Armas, JM, Öztürk, MM, Simon, M, O’Neill, E, Bryce-Smith, S, Harrison, J, Atkins, G, Corrochano, S, Stewart, M, Teboul, L, Acevedo-Arozena, A, Fisher, EMC, Cunningham, TJ. Generation and analysis of innovative genomically humanised knock-in SOD1, TARDBP (TDB-43), and FUS mouse models. ISCIENCE (2021), https://doi.org/10.1016/j.isci.2021.103463